
2026 年初,印度西孟加拉邦尼帕病毒确诊病例再现,医护人员感染事件引发全球警惕。这种致死率高达 40%-75% 的人畜共患病毒,至今无特效药与疫苗,防控核心全靠 “早检测、严防护”。对于 IVD、器械及防护产品企业而言,这既是紧急市场需求,更是一场合规能力的大考。本文结合各国官方最新监管要求,拆解病毒防控全链条的合规关键,为企业打通全球市场提供清晰指引。

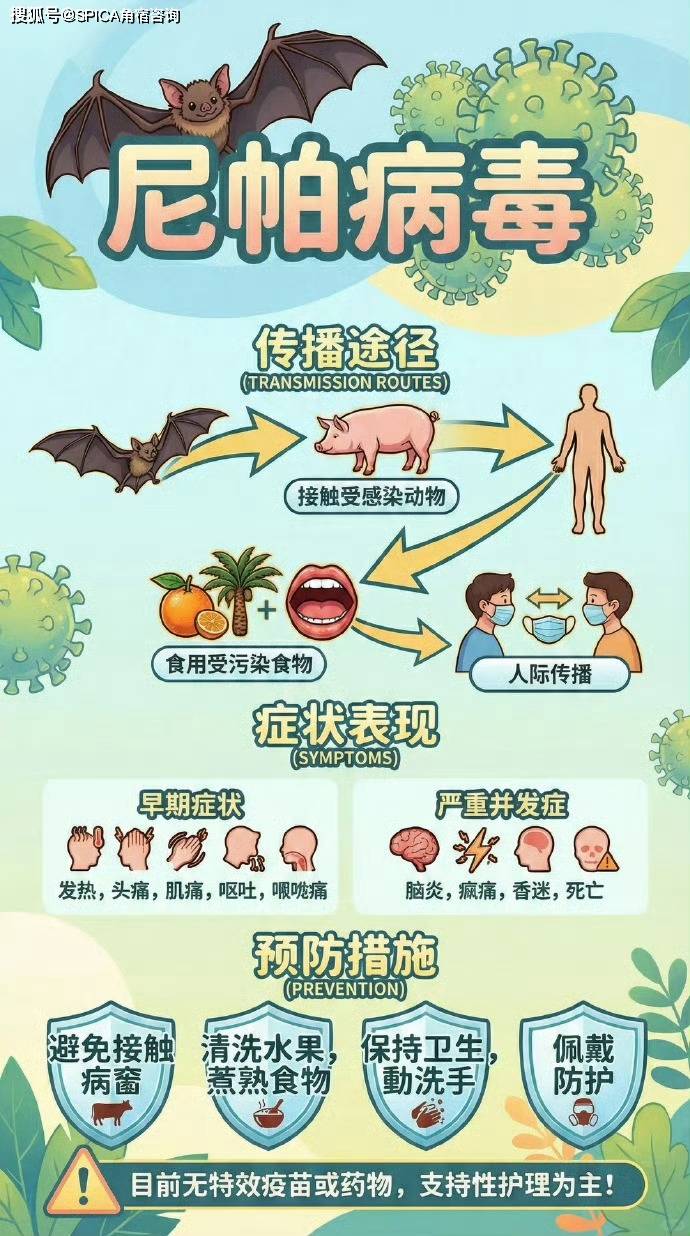
要精准布局合规策略,必先明晰尼帕病毒的传播特性与核心防控产品的定位。据世界卫生组织(WHO)最新通报,尼帕病毒主要通过蝙蝠、猪等动物分泌物污染的食物传播,人际传播多发生在医疗场景,潜伏期最长可达 45 天,隐性传播风险极高。2024 年 12 月,中国已将其列入《监测传染病目录》,2026 年进一步升级入境防控措施,足见其公共卫生风险等级。
核酸检测试剂:作为确诊 “金标准”,核心要求高灵敏度与特异性,适配口岸检疫、疾控中心批量筛查,需满足各国对检测限(LoD)、无交叉反应的严格要求;
快速检测产品(抗原 / 抗体):主打基层医疗与现场筛查,核心要求操作简便、15-30 分钟出结果,无需专业实验室设备,是社区防控的关键工具;
检测设备:全自动核酸提取检测系统、高通量测序平台等,可降低生物安全风险,成为省级疾控与大型实验室的核心采购标的。
结合传播途径与风险场景,官方明确分级防护标准,不同产品对应不同合规要求:
基础防护:N95 及以上级别呼吸防护器、平光镜(防范飞沫入眼),适配日常出行与低风险场景;
高级防护:一次性防水隔离衣、双层手套、护目镜,针对医疗等高风险环境,对防水性、防气溶胶性能要求极高;
环境防控:含氯消毒液、75% 酒精等消杀产品,需符合各国公共卫生消毒标准,是环境防疫的重要支撑。
不同国家与地区的监管体系差异显著,企业需精准匹配目标市场的合规要求,避免踩入监管陷阱。以下结合中国 NMPA、美国 FDA、欧盟监管机构及疫区重点国家最新政策,梳理核心合规要点。
中国:已将尼帕病毒检测试剂纳入应急审批绿色通道,大幅缩短审批周期。企业需提交产品性能验证报告、临床试验总结、质量管理体系证明等资料,获批后可纳入口岸检测与疾控采购目录;
美国:依据 2026 年 1 月 FDA 发布的最新指南草案,针对新兴病原体 IVD 产品,可通过紧急使用授权(EUA)快速获批,核心需提供至少 30 份阳性与 30 份阴性样本的验证数据,无症状筛查产品需满足 98% 以上的阴性百分比一致性(NPA);
欧盟:需符合 IVDR(体外诊断医疗器械法规)要求,尼帕病毒检测试剂多归为中高风险类别,需通过公告机构审核,提交完整技术文件与临床性能数据,参考猴痘检测试剂 IVDR 认证的核心标准,确保检测准确性与可靠性;
疫区国家(如印度):实施临时注册机制,需提供产品质量合格证明、原产国自由销售证书,核心考核与本地流行毒株的匹配性。
中国:医用防护口罩需符合 GB 19083-2010 标准,医用防护服需符合 GB 19082-2009 标准,企业需获得医疗器械注册证(或备案凭证),进口产品需指定国内代理人并通过 NMPA 注册;
美国:N95 口罩需通过 NIOSH 认证,医用防护装备可申请 FDA 510 (k) 许可或 EUA 授权,核心考核过滤效率、液体阻隔性能等指标;
欧盟:按 PPE(个人防护装备)法规分级认证,口罩多为 II 类或 IIR 类,防护服需符合 EN 14126 标准,需通过公告机构检测并标注 CE 标志及唯一标识;
东南亚疫区:对进口防护产品实施严格抽检,需提供符合当地标准的检测报告,部分国家要求额外完成本地代理人备案。

美国:需通过 EPA(环境保护署)注册,常规流程需提交产品化学、毒理学等数据,周期约 12-24 个月,中小企业可申请简化流程与费用减免;
欧盟:需符合 BPR(生物杀灭剂法规),实行 “活性物质批准 + 成品注册” 两步走机制,需承担数据共享费用,整体周期长达 3-5 年;
中国:需完成卫生安全评价并备案,提交微生物检测报告与安全性评估资料,标签需明确标注有效成分与使用范围。
企业在尼帕病毒防控产品合规中,常面临 “审批慢、标准乱、后续风险高” 等问题,需建立全流程风险防控体系,精准弥补合规漏洞。
新兴病原体产品的核心优势在于 “快”,企业需善用各国应急通道:申请美国 EUA 时,严格按 FDA 指南准备样本验证数据,确保阳性百分比一致性(PPA)不低于 95%;进入中国市场时,优先走应急审批绿色通道,提前梳理技术文件以缩短审核周期。同时,需提前明确产品分类,避免因分类错误导致合规路径偏差。
不同国家的技术标准差异显著,需逐一精准匹配:出口欧盟的防护服需满足 EN 14126 标准的防水、防气溶胶要求;申请 EPA 注册的消杀产品,需严格遵循标签标注规范,禁止未经证实的功效宣称;IVD 试剂需按目标国要求补充样本验证,疫区国家需额外提供本地毒株的适配性数据。
针对新兴病原体产品的特殊性,需重点强化资料完整性:FDA EUA 申请需明确标注样本类型(天然标本优先,掺假样本需满足特定比例要求);欧盟 BPR 注册需提前规划活性物质评估,参与联合注册以分摊费用;猴痘试剂 IVDR 认证的成功经验显示,完整的临床性能数据与风险管理报告是审批关键。
获证并非终点,需建立法规动态监测机制:美国 EUA 产品需按要求提交后续性能数据,欧盟 IVDR 认证产品需配合公告机构的定期审查;EPA 与 BPR 均有定期复审要求,需及时更新物质清单与风险评估报告;中国应急获批产品需按要求完成后续补充临床试验,确保长期合规。

尼帕病毒防控产品的合规之路,考验的是对全球法规的精准解读与全流程把控能力。SPICA 角宿咨询依托深厚的行业积累与全球资源,为 IVD、器械及防护产品企业提供全链条合规支持:
精准研判:深度解读 WHO 及各国最新监管政策,结合疫区防控动态,为企业提供目标市场合规可行性评估,明确产品分类、审批路径及核心难点;
定制方案:针对检测试剂、防护装备、消杀产品的不同特性,以及中国、美国、欧盟、疫区国家等不同市场,定制专属合规方案,优先利用应急通道缩短获批周期;
漏洞弥补:从技术文件撰写、性能验证指导,到注册申报、现场核查协调,全程由资深合规专家跟进,精准解决样本验证、标准对标、资料补正等核心问题;
全球协同:依托沙特、加拿大、欧洲等海外分公司资源,提供本地代理人、检测对接、标签审核等服务,破解跨国沟通与监管壁垒。
在尼帕病毒防控的紧急战场上,合规速度直接决定市场先机。欢迎绿泡泡添加咨询合规方案:SPICA-15,SPICA 角宿咨询以专业、高效的合规服务,助力企业在守护全球公共卫生安全的同时,顺利打通全球市场通道,实现商业价值与社会价值的双重提升。返回搜狐,查看更多


